- Hobbies include neighborhood walks and playing bingo
- Married, mother of 3, with 4 college-aged grandchildren
- Motivated and active participant in her treatment
- Married, father of one, with 2 grandchildren
- Presents with back and bone pain, and high symptom burden
- Highly limited in mobility and self-care

- Lower back pain
- Diagnosed with mild dementia 3 years ago (current MMSE score of 22)
ECOG PS=Eastern Cooperative Oncology Group Performance Status; ISS=International Staging System; MMSE=Mini-Mental State Examination.
*Cytogenetic risk was based on fluorescence in situ hybridization (FISH) or karyotype analysis. High-risk cytogenetics were defined as having at least one of the following abnormalities: t[4;14], t[14;16], del[17p], t[14;20], gain[1q21], amp[1q21].3
- Good support system: not married but in a steady relationship past 20 years
- At times, struggles to stay adherent to treatment
Presents with fatigue due to multiple myeloma and reduced lung capacity- Presented with diabetes
- Requires assistance to get around, especially up and down stairs
- Married, with 3 adult children
- Enjoys spending time with family and friends
- Looks forward to taking on new projects at work
Cytogenetic risk: Standard*Presents with:
- Mild cardiac disease†
- Mild persistent asthma‡
- Chronic kidney disease with moderate renal dysfunction§


- Primary endpoint was progression-free survival (PFS) based on International Myeloma Working Group (IMWG) criteria2
- Key secondary endpoints included percentage of patients with complete response (CR), very good partial response (VGPR) rate, minimal residual disease (MRD)–negativity rate (next-generation sequencing [NGS]; 10-5), overall response rate (ORR), overall survival (OS), duration of response, and safety2
- Baseline demographics and disease characteristics were similar between the 2 treatment groups1


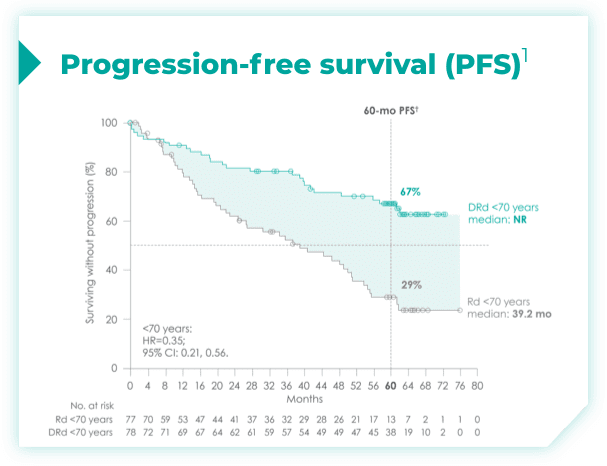
- ~50% of patients were 75 years old or older2
- The trial also included patients with various ECOG PS, cytogenetic profiles, and ISS disease stages2






- ISS disease stage: II
- ECOG PS: 1
DARZALEX® + Rd is the only anti-CD38 triplet regimen with proven long-term overall survival results.1


(DRd: 95% CI: 61,71; Rd: 95%: 47,59)2*†
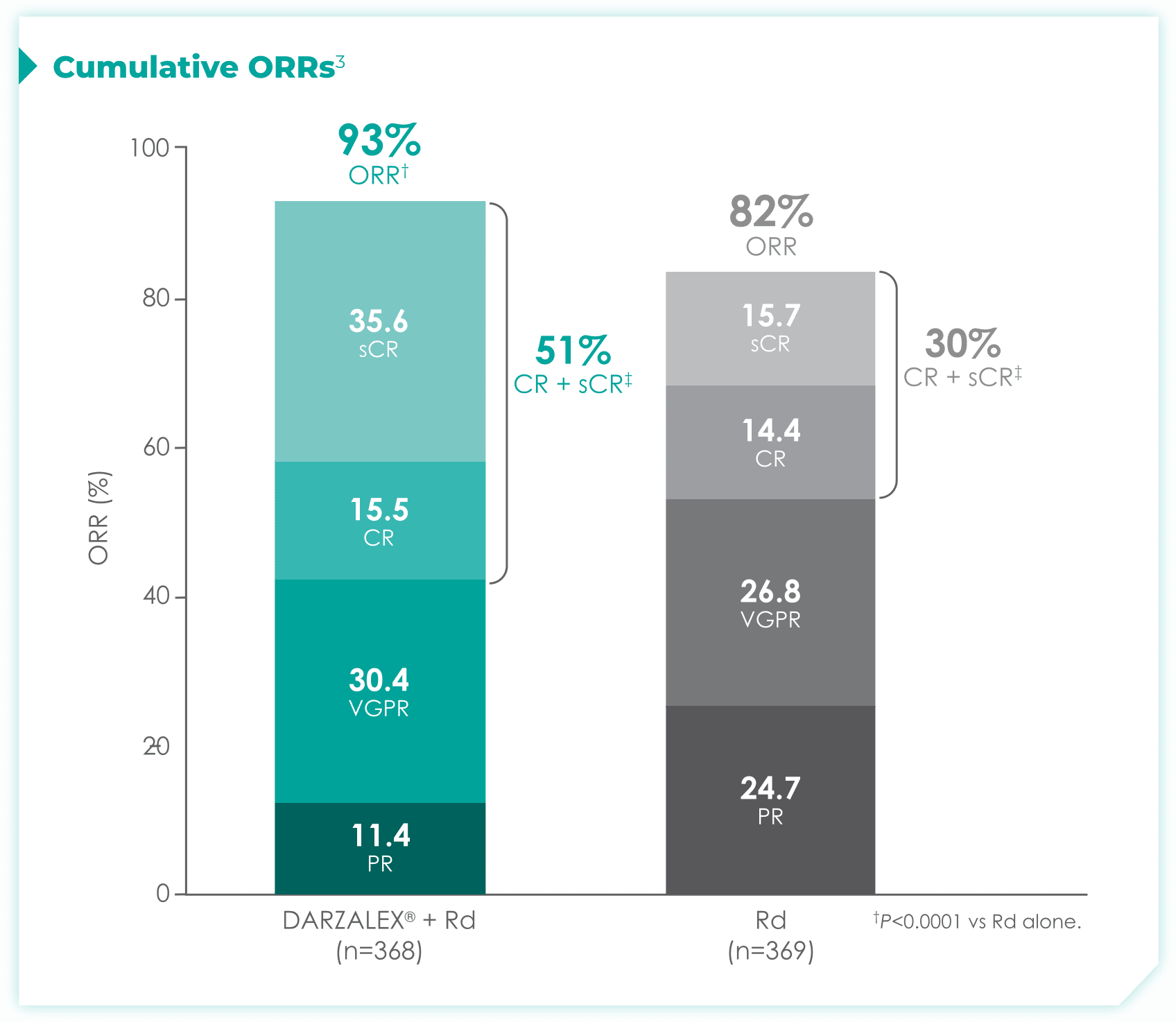
with Rd alone3*
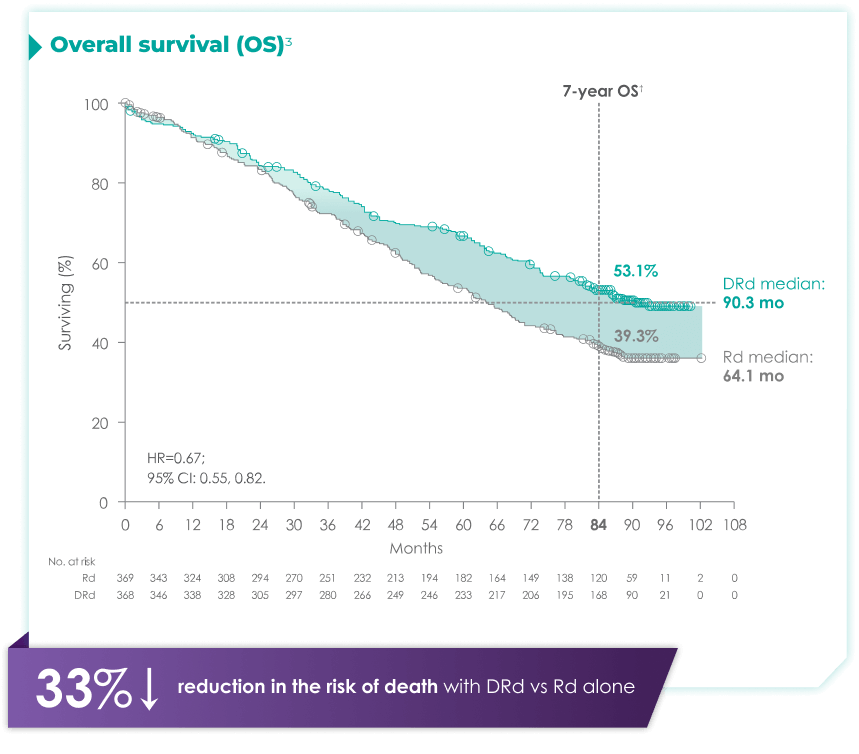
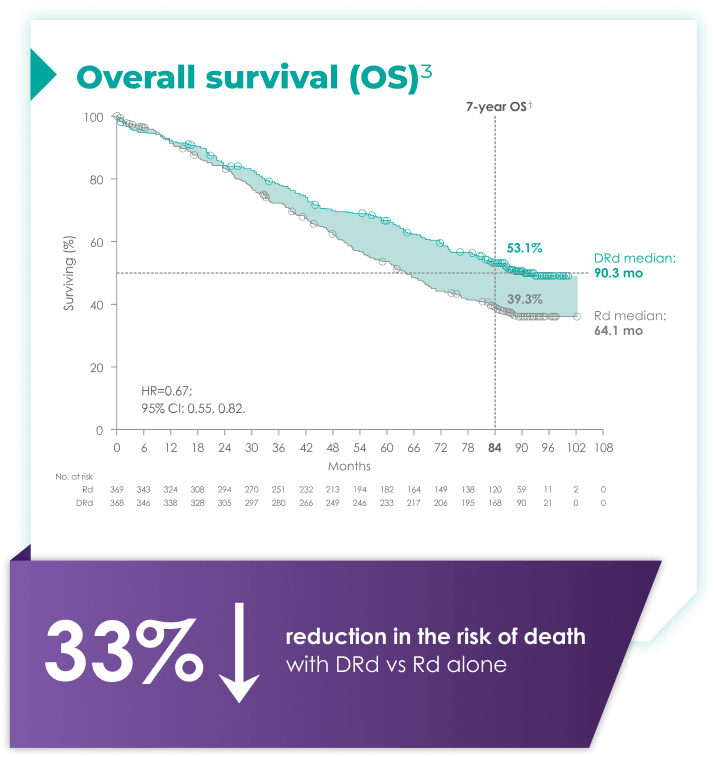
Rd=lenalidomide (R) + dexamethasone (d).
- ISS disease stage: II
- ECOG PS: 1
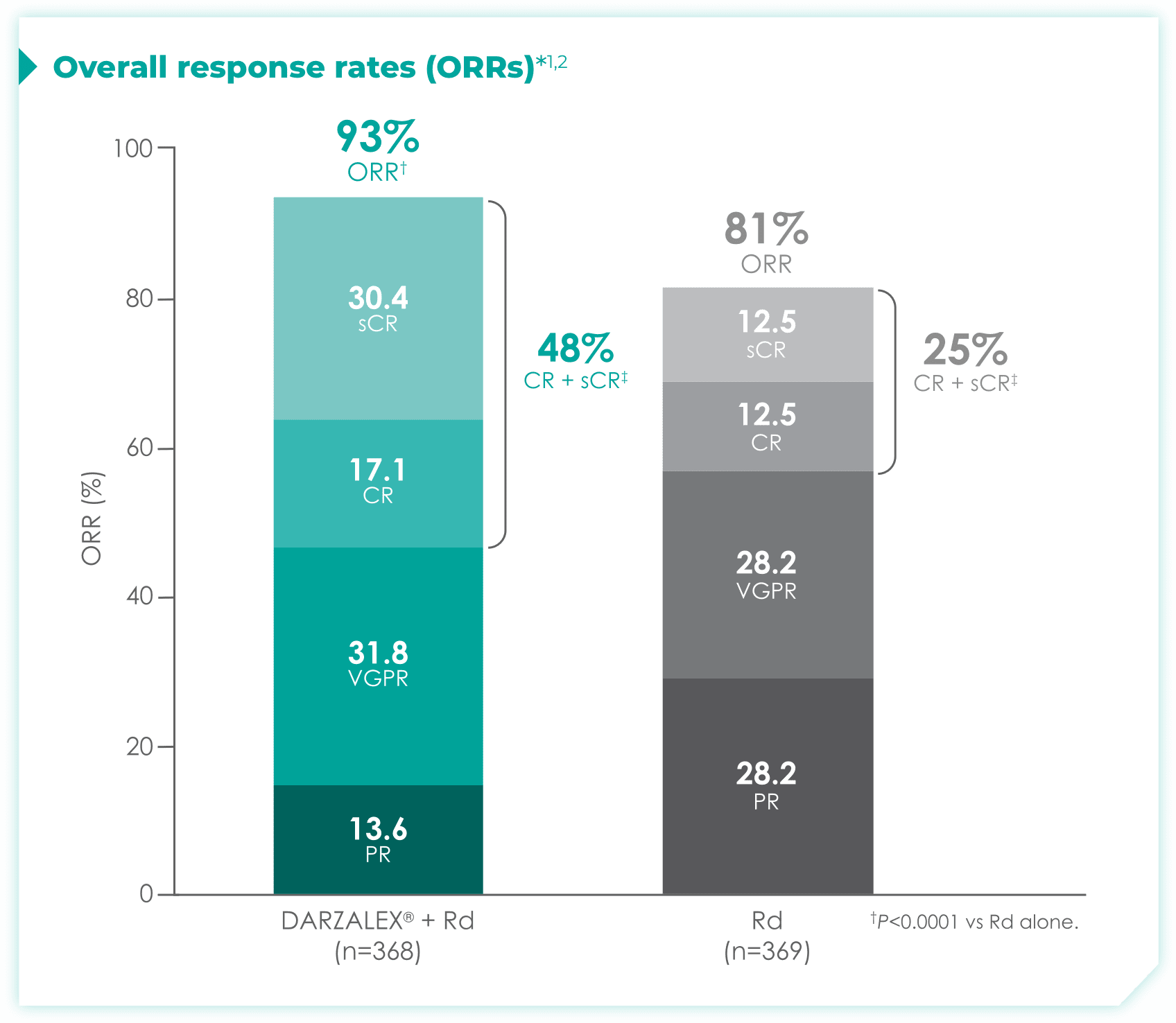

DRd nearly doubled the number of patients who achieved CR† or better vs Rd alone.1
- More than doubled sCR‡: 30% with DRd vs 13% with Rd alone
Median duration of response had not yet been reached with DRd vs 34.7 months (95% CI: 30.8, not estimable) for Rd alone.1
Median time to response was 1.05 months with DRd (range: 0.2-12.1 months) and with Rd alone (range: 0.3-15.3 months).1
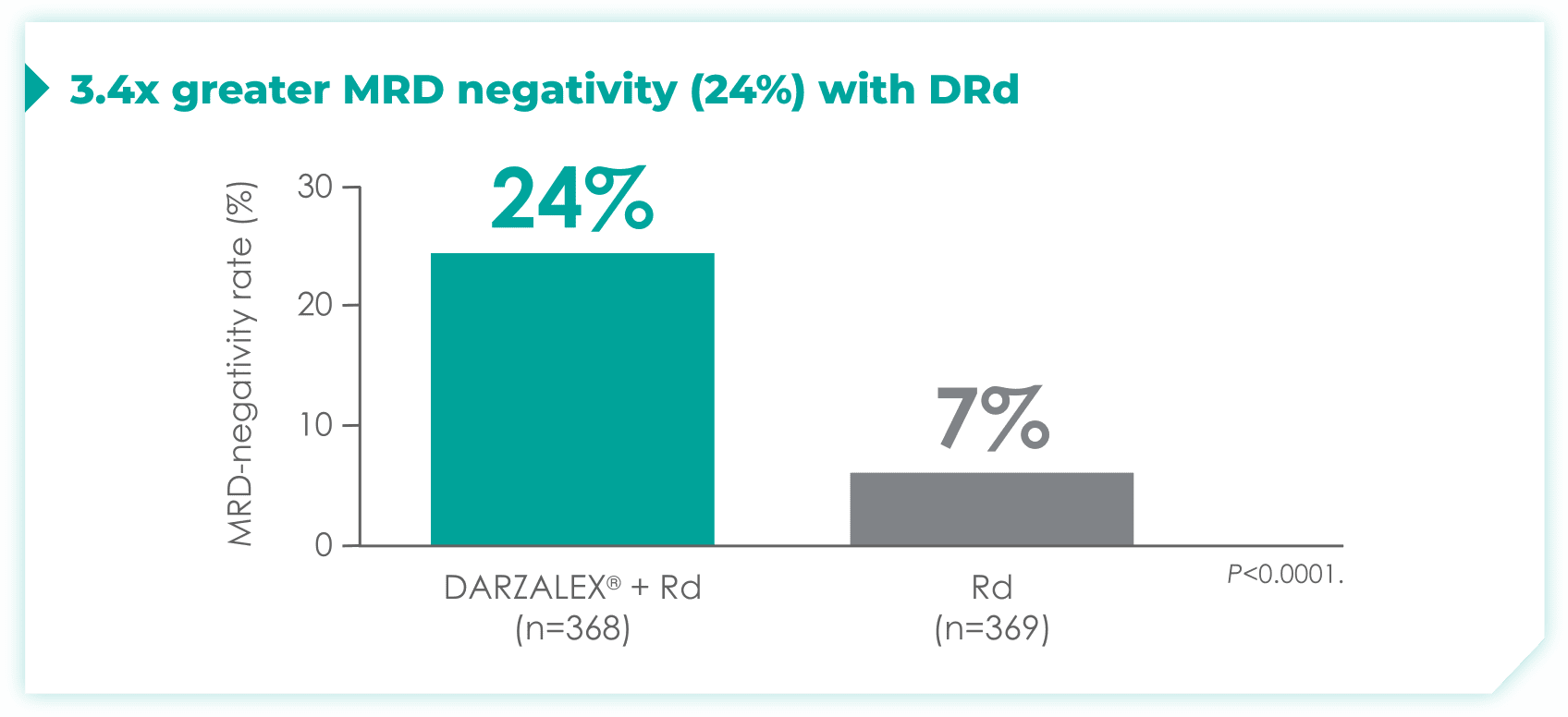

- 24% of patients were minimal residual disease (MRD)-negative with DRd (95% CI: 19.9, 28.9)
- 7% of patients were MRD-negative with Rd (95% CI: 4.9, 10.5)1

1.7x more patients receiving DRd achieved CR or better vs Rd alone
- More than doubled sCR‡: 36% with DRd vs 16% with Rd alone








- Discontinuation rates due to any adverse event: 7% with DRd vs 16% with Rd2
- Infusion-related reactions (IRRs) with DRd occurred in 41% of patients; 2% were Grade 3 and <1% were Grade 41
- IRRs of any grade or severity may require management by interruption, modification, and/or discontinuation of the infusion1
- Most IRRs occurred during the first infusion1


- Cumulative Grade 3/4 infection rates were 43% for DRd vs 30% for Rd3†
- Cumulative rates of discontinuation due to TEAEs were 15% for DRd vs 24% for Rd3†
- Hematologic adverse events included in the follow-up analyses are investigator-reported TEAEs and not investigator-reported treatment-emergent laboratory abnormalities
- The primary reason for discontinuation in both the DRd and Rd groups was progressive disease (32.7% and 38.6%, respectively)
- A lower proportion of patients in the DRd group vs the Rd group discontinued study treatment due to adverse events (AE) (16.5% and 25.8% respectively)


- For 37% of patients, IRRs (any grade) occurred with the Week 1 (16 mg/kg) infusion; for 2% of patients, with the Week 2 infusion; and cumulatively, for 6% of patients with subsequent infusions
- Median time to onset of an IRR was 1.5 hours (range: 0–73 hours)
- Incidence of infusion modification due to reactions was 36%
- DARZALEX® can cause severe IRRs. Severe IRRs included bronchospasm, hypoxia, dyspnea, hypertension, tachycardia, headache, laryngeal edema, pulmonary edema, and ocular adverse reactions, including choroidal effusion, acute myopia, and acute angle closure glaucoma. Signs and symptoms may include respiratory symptoms, such as nasal congestion, cough, throat irritation, as well as chills, vomiting, and nausea. Less common symptoms were wheezing, allergic rhinitis, pyrexia, chest discomfort, pruritus, hypotension, and blurred vision
- Ocular adverse reactions, including acute myopia and narrowing of the anterior chamber angle due to ciliochoroidal effusions with potential for increased intraocular pressure or glaucoma, have occurred with DARZALEX® infusion. If ocular symptoms occur, interrupt DARZALEX® infusion and seek immediate ophthalmologic evaluation prior to restarting DARZALEX®


- Type and screen patients before starting DARZALEX®
- Inform blood banks when a patient is on DARZALEX®
- Identify any blood samples of patients treated with DARZALEX®
- Ask patients to tell other healthcare professionals that they've taken DARZALEX®
Brochure on daratumumab and serological testing
Identification card to give to patients taking daratumumab




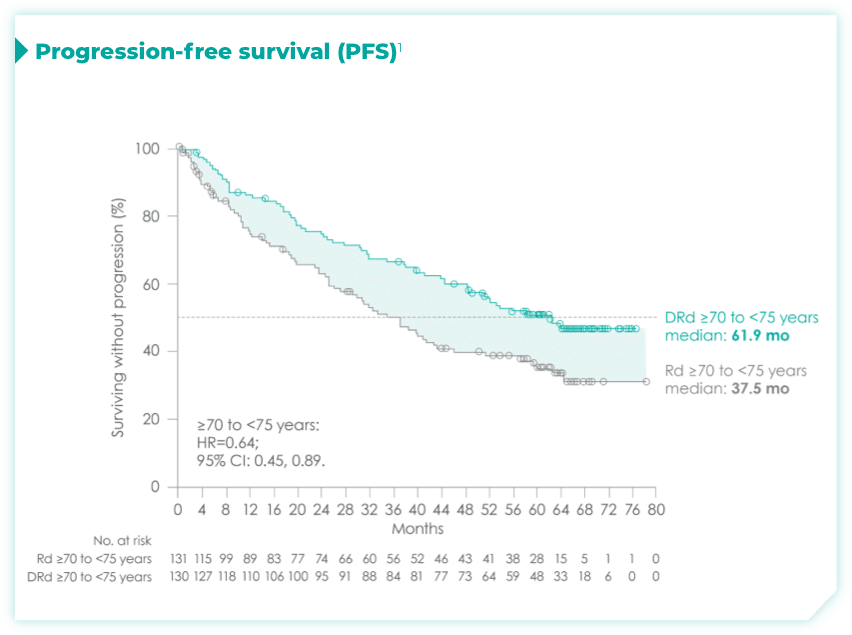
- Among patients with revised standard cytogenetic risk (0 HRCA), median progression-free survival (mPFS) was not reached with DRd vs 35.1 months with Rd alone (HR=0.50; 95% CI: 0.37, 0.66)1*






- ISS disease stage: III
- ECOG PS: 2
Do you have patients in your practice who may be appropriate for frontline DRd?
Dr. Shain reviews key considerations for treating patients like Luis


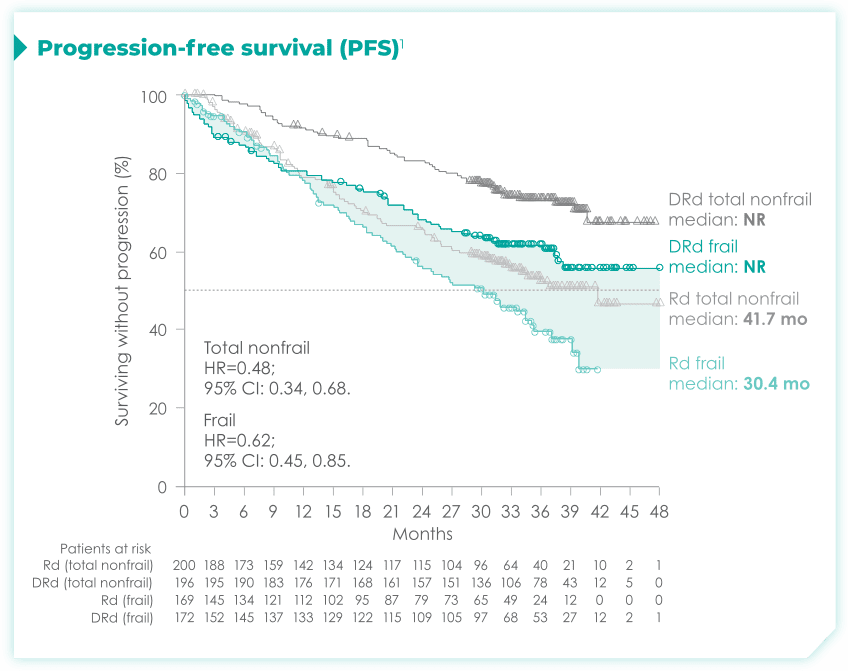
- The median age in frail subgroup was 77 years (range: 57-90 years), with 88% of patients having ECOG PS ≥11
- CCI was calculated based on retrospective review of each patient’s medical history1





- ISS disease stage: II
- ECOG PS: 2
Do you have patients in your practice who may be appropriate for frontline DRd?
Dr. Birhiray reviews key considerations for treating patients like Samuel



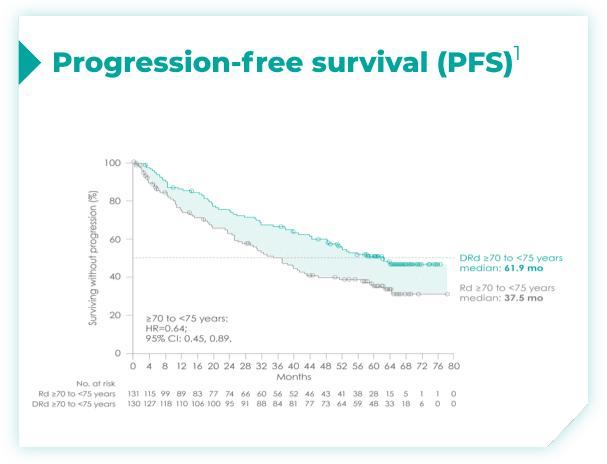
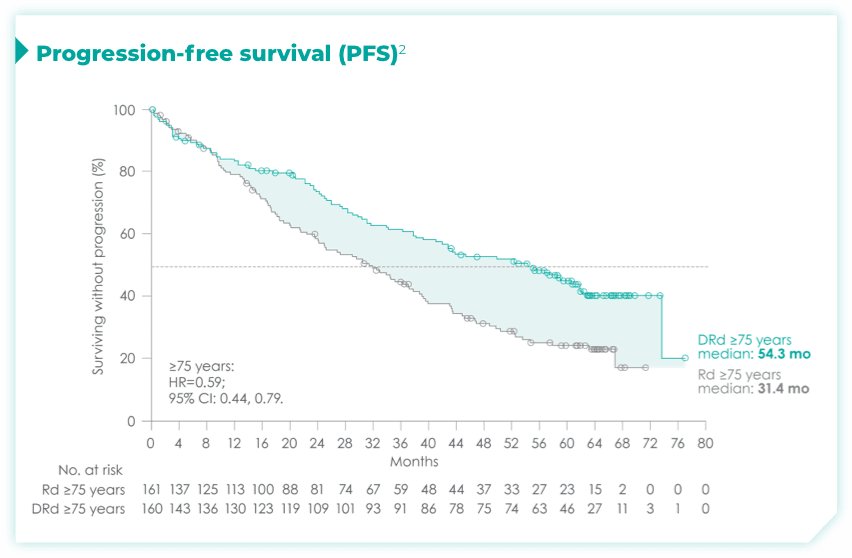
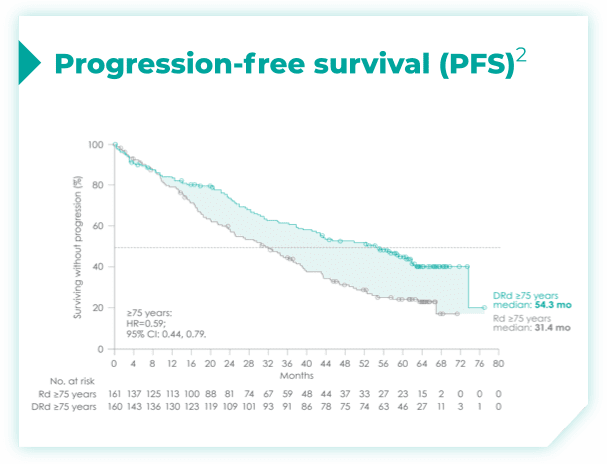


- ISS disease stage: I
- ECOG PS: 1
References:
- DARZALEX® [Prescribing Information]. Horsham, PA: Janssen Biotech, Inc.
- Facon T, Kumar SK, Plesner T, et al. Daratumumab, lenalidomide, and dexamethasone versus lenalidomide and dexamethasone alone in newly diagnosed multiple myeloma (MAIA): overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22(11):1582-1596.
- Moreau P, Facon T, Usmani SZ, et al. Daratumumab plus lenalidomide and dexamethasone (D-Rd) versus lenalidomide and dexamethasone (Rd) in transplant-ineligible patients with newly diagnosed multiple myeloma (NDMM): clinical assessment of key subgroups of the phase 3 MAIA study. Poster presented at: 64th American Society of Hematology (ASH) Annual Meeting & Exposition; December 10-13, 2022; New Orleans, LA.